An Unusual Case of vanishing bone disease

Dr Jarrad Stevens and colleagues have reported a case of vanishing bone disease which has been published in BMJ Case Reports.The case history of the woman with the unusual diagnosis of Gorham-Stout disease goes like this.
A previously healthy 44-year-old female presented with an acute history of increasing pain and reduced range of movement in her left shoulder. MRI scan revealed an infiltrative lesion in the proximal humerus, with a degree of cortical thinning and soft tissue involvement; however, initial biopsy provided no definitive diagnosis. Due to the suspicion of underlying malignancy, additional biopsies were organized. A second biopsy provided no diagnosis; however, a third biopsy taken 2 months after presentation revealed a benign vascular lesion with callus formation. This was consistent with the suspicion of our radiologist that a pathological fracture of the proximal humerus had been sustained following a minor fall.
Figure 1Radiograph of left humerus and shoulder at presentation.
Twelve months after presentation, with continued pain and swelling in the arm, further investigations were carried out. Radiographs demonstrated a second pathological fracture of the left proximal humerus, with intramedullary and subcortical radiolucent foci (figure 2). Contrast MRI scans and angiogram showed a more extensive vascular lesion than previously seen with no sign of arteriovenous malformation. Bone biopsy showed a highly vascular lesion with lymphovascular exudate and no obvious solid lesional tissue. Bone and soft tissue samples showed prominent osteoclastic bone resorption (figure 3). CT chest, abdomen and pelvis revealed no sign of malignancy. At that point, bone scan demonstrated isolated humerus involvement. However, subsequent radiographs taken 1 month later revealed rapidly spreading disease, with near-complete resorption of the humerus, and suspicion of disease spread to the ulnar (figure 4). At this point, with the histological and radiological findings, a diagnosis of Gorham-Stout disease (GSD) was considered, with a second opinion sought from the multidisciplinary team in Birmingham. A consensus diagnosis of rapid and multifocal GSD was made. The most recent X-ray is seen in figure 5.
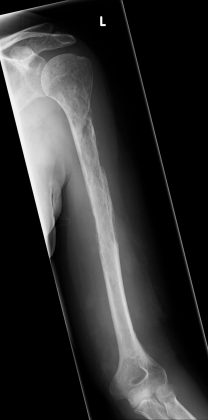 Figure 2 Radiograph of left humerus 12 months after initial presentation.
Figure 2 Radiograph of left humerus 12 months after initial presentation.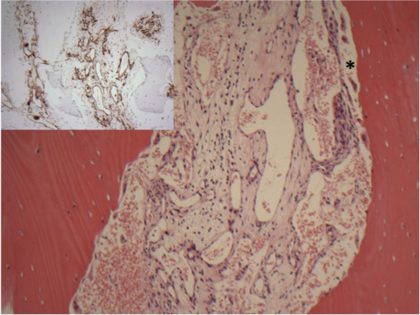 Figure 3
Figure 3Biopsy of bone shows the marrow space to be replaced by thin-walled vessels of varying calibre. There is prominent osteoclastic bone resorption.* Main panel: H&E stain, original magnification ×10. Inset: CD31 immunohistochemistry, original magnification ×10.
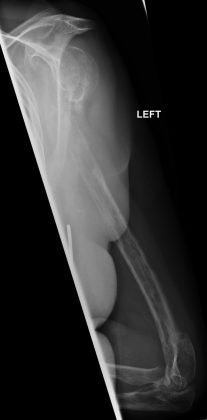 Figure 4
Figure 4Radiograph of left humerus and elbow 15 months after presentation. Rapidly progressing and multifocal Gorham-Stout disease.
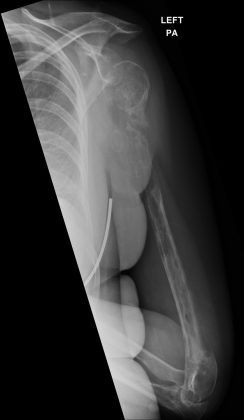 Figure 5
Figure 5Radiograph of left humerus 18 months after presentation. PA, posterioranterior.
GSD or ‘vanishing bone’ disease is a rare disorder, characterized by the destruction of osseous matrix due to the aggressive proliferation of non-neoplastic vascular and lymphatic tissue, similar to a haemangioma or lymphangioma.1 Only 64 cases have been reported in the literature, 8 of which have involved the humerus. While the condition is considered benign, the prognosis is uncertain, with potentially fatal complications that include disease spread to the vertebrae, leading to pleural effusion and quadriplegia. The progressive bone loss can lead to complete bone resorption. Associations have been proposed between massive osteolysis following trauma or as a result of increased osteoclast and interleukin 6 activity. In spite of this, the definitive aetiology and pathology of the disease remain unknown. Due to its rarity, diagnosis is challenging and first requires the exclusion of more common disorders including neoplastic, inflammatory, infectious and endocrine disease. A presentation can include pain, functional impairment and swelling, although asymptomatic cases have been described. An x-ray may show atrophy, bone tapering, and pathological fractures. The gold standard for diagnosis is a biopsy demonstrating angiomatous tissue.2 This is an integral component of the diagnostic criteria for GSD proposed by Heffeze et al3:
minimal/no osteoblastic response and absence of dystrophic calcification
positive biopsy for angiomatous tissue
absence of cellular atypia
evidence of local progressive osseous resorption
non-expansile, non-ulcerative lesion
absence of visceral involvement
osteolytic radiographic pattern
negative hereditary, metabolic, neoplastic, immunological or infectious aetiology.
Treatment options remain limited and have varying success; some positive reports have been made after the use of multimodal therapy (radiation therapy, bisphosphonates and surgical resection). Ultimately, this is a challenging disease where evidence-based management remains lacking.
Footnotes
Contributors All authors contributed to the paper. JS and HF contributed to the literature review, case report write-up and draft manuscript. JTP conceived the notion that this rare disease would be of importance to other surgeons, edited and checked the manuscript and references.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
For more details click on the following link : doi:10.1136/bcr-2017-224061
Disclaimer: This site is primarily intended for healthcare professionals. Any content/information on this website does not replace the advice of medical and/or health professionals and should not be construed as medical/diagnostic advice/endorsement or prescription. Use of this site is subject to our terms of use, privacy policy, advertisement policy. © 2020 Minerva Medical Treatment Pvt Ltd